
期刊:Science China-Life Sciences
影响因子:8.0
生命科学研究的显微镜化证明,基于细胞群的方法可能不适用于某些领域的研究,如肿瘤的异质性和早期胚胎发展作为回应,2009年单细胞转录组测序技术的引入标志着一项重大进步(Tang,2009)。在此基础上,Navin等人(2011)于2011年将全基因组扩增(WGA)与高通量测序相结合,引入了单细胞全基因组测序技术(scWGS)。这种创新的方法解决了获取组织样本中不同细胞之间异质性信息的挑战,当传统测序由于样本量小而不切实际时,可以研究单个细胞。通过在单细胞水平上的DNA测序,scWGS为研究单个细胞的行为和机制提供了一个新的维度。scWGS的应用已经扩展到各个研究领域,包括神经科学、种系进化、器官发生、肿瘤学、临床诊断、免疫学、微生物学、胚胎发育和产前遗传学诊断。认识到其潜力,scWGS被《自然方法》杂志强调为2013年最受期待的技术之一。
scWGS的发展确实为研究人员在单细胞水平上探索细胞间异质性,探索单核苷酸变异、短插入或缺失、拷贝数变异等各个方面开辟了途径。该技术已被证明对研究具有生物学或临床意义的罕见细胞的基因组特别有价值,包括循环肿瘤细胞和用于第三代体外受精植入前遗传学诊断/筛选的细胞。scWGS过程通常包括三个主要步骤:单细胞分离、单细胞全基因组扩增(scWGA)和扩增产物的测序和分析。这一过程中的关键挑战是有效地扩增单个细胞的基因组,获得足够的材料用于下游分析,同时最大限度地减少扩增偏倚、基因组丢失、突变和嵌合体等人工产物。解决这些挑战对于确保从单个细胞中获得的遗传信息的准确性和可靠性至关重要。
在本节中,首先将重点介绍scWGA技术的进展。随后详细介绍了几个突出的scWGA化学物质,阐明了它们的关键生化反应策略。然后,我们的重点转向高通量scWGS方法,这使大规模的肿瘤细胞基因组并行测序成为可能。这种方法为显著拓宽肿瘤内特征的范围提供了机会。scWGA和高通量scWGS技术发展的关键里程碑如图5所示。最后,我们总结了scWGS在生物医学领域的最新实际突破。本概述概述了将单细胞基因组测序应用于临床研究的愿景,强调了其对推进我们对生物过程和疾病机制的理解的潜在影响。
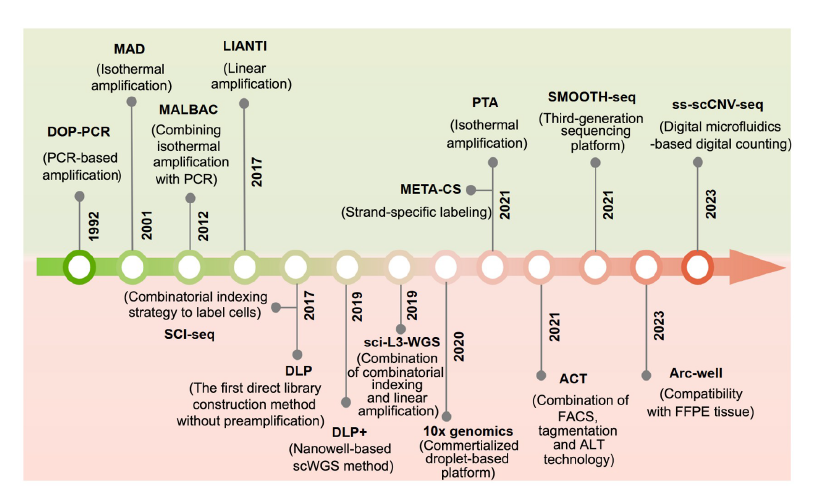 Fig 5. 单细胞全基因组扩增和测序技术的发展概况。图表的上半部分显示了标记的主要事件scWGA和低通率scWGS技术的发展,以及吞吐量的发展
Fig 5. 单细胞全基因组扩增和测序技术的发展概况。图表的上半部分显示了标记的主要事件scWGA和低通率scWGS技术的发展,以及吞吐量的发展
ScWGA方法
由于单个细胞中的DNA含量有限(约6pg/细胞),没有达到测序仪的检测要求,因此必须在测序前放大单个细胞中微量的全基因组DNA。该扩增过程的目的是生成高覆盖率的全基因组,确保后续高通量测序中准确和全面的测序结果。随着时间的推移,WGA技术发生了重大变化,以满足这些需求。值得注意的方法包括简并寡核苷酸引物聚合酶链反应(DOP-PCR)(Telenius,1992),多重置换扩增(MDA)(Dean,2001),以及多重退火和基于循环的扩增循环(MALBAC)(Zong,2012)。随后的创新,如通过转座子插入的线性扩增(LIANTI)(Chen,2017),单链测序利用微流控反应器(SISSOR),初级模板定向扩增(PTA)(Gonzales -Pena,2021)和互补链的多重末端标记扩增(META-CS)(Xing,2021)进一步扩展了WGA工具包。表1给出了这些方法的一般特征的概述。在下面的章节中,将回顾几种主要的WGA方法,重点关注它们的覆盖范围、均匀性和准确性。简而言之,放大性能没有明显的赢家,每种策略根据重要的参数都有优势。
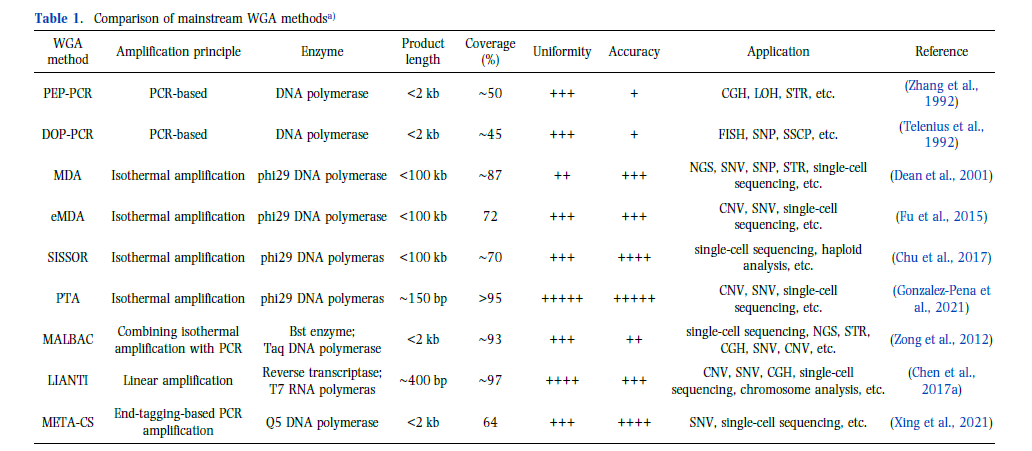
表1:CGH,比较基因组杂交;LOH,杂合性缺失;STR,短序列重复;FISH,荧光原位杂交;SNP,单核苷酸多态性;SSCP,单链构象多态性;NGS,第二代测序;CNV,拷贝数变异;SNV,单核苷酸变异
PCR扩增
基于PCR的WGA方法有多种形式,包括引物延伸预扩增PCR(PEP-PCR)(Zhang,1992)、DOP-PCR(Teleneius,1992)、标记随机引物PCR(T-PCR)(Grothues,1993)和连接介导PCR(LM-PCR)(Klein,1999)。这些技术对于从单个细胞中获得基因组DNA的扩增、满足特定的研究目标以及作为scWGA技术的模型至关重要。最早使用的scWGA技术之一是PEP-PCR,它后来被更流行的DOPPCR所取代。这些技术导致了商业试剂盒的发展,如PicoPLEX WGA试剂盒(Rubicon Genomics,美国)和GenomePlex单细胞全基因组扩增试剂盒(Sigma-Aldrich,美国)。
DOP-PCR的原理是利用部分随机引物对模板基因组DNA进行两步PCR扩增(Telenius et al.,1992)。简并引物由中间随机的6个碱基序列组成,两侧各有固定序列。引物3’端较短的ATGTGG序列在基因组DNA中具有极高的分布频率,指导了初始低温退火步骤,并确定了偏向于特定序列的扩增起始位点。中间的6个简并碱基产生了46个不同的序列,在退火过程中,其中一个或多个简并碱基与3’端特异性碱基同时与模板DNA结合,提高了引物的结合效率。PCR扩增分为两个步骤:前几个周期(3-5个)涉及低温退火(例如,30°C),然后在高温下进行链延伸。在第二步中,在更高的退火温度(62°C)下,使用针对5’固定序列的引物进行进一步的扩增。
DOP-PCR扩增的效率取决于引物浓度和聚合酶活性。在低退火温度下,引物可以结合多个基因组位点,产生扩增产物几乎覆盖整个基因组。DOP-PCR是基于PCR的WGA中最突出的一种代表性方法,可扩增单细胞的人类基因组DNA,用于肿瘤异质性分析、拷贝数变异评估和非整倍体检测(Knouse,2014;McConnell,2013;Navin,2011)。然而,DOP-PCR通常产生较低的基因组覆盖率(通常低于10%)(Navin,2011),这是一个与PCR扩增的指数性质相关的特征。此外,PCR扩增反应具有较高的碱基错配率,由于假阳性率升高,不太适合检测单核苷酸变异。
恒温扩增
MDA是等温scWGA方法中最具代表性的方法,最初由Dean等人(2001)开发。MDA在等温条件下运行,采用了一个6-碱基对的随机引物,随机退火到基因组上,启动了一个由phi29 DNA聚合酶催化的链置换扩增反应。通过置换产生的单链序列可以通过随机引物退火进行随机扩展,从而形成多分支扩增结构。由于phi29 DNA聚合酶具有强大的DNA合成能力,合成的DNA片段长度通常为50-100kb。此外,phi29 DNA聚合酶的高复制保真度,由于其3’→5’外切酶和校对活性,其错误率约为每108个1个核苷酸,使得MDA适用于精确的单核苷酸变异(SNV)。这一特性导致了其在单细胞基因组谱系追踪中的应用(Lodato,2015)。此外,与最初的基于PCR的方法相比,MDA提供了显著更高的基因组覆盖率。然而,MDA的一个缺点是其指数扩增过程,类似于DOP-PCR,它引入了序列依赖的偏倚,阻碍了覆盖的均匀性。值得注意的是,MDA的序列依赖偏倚在从一个细胞到另一个细胞的基因组中不能一致地重复,这使得拷贝数变异(CNV)测量的噪声和归一化效率较低。
为了解决放大偏差和增强均匀性和覆盖率,各种改进的等温放大技术,包括乳剂MDA(eMDA)(Fu,2015),数字液滴MDA(ddMDA)((Sidore,2016),TruePrime等,2016)、SISSOR(Chu,2017)和PTA(Gonzalez-Pena,2021)已经基于MDA技术开发。eMDA和ddMDA都涉及到将扩增过程分散到数百万个小液滴中,旨在提高扩增的均匀性和纠正偏置。TruePrime技术用一种名为TthPrimPol的独特DNA引物替代MDA方法中的N6引物,以增强扩增均匀性。SISSOR通过将同源染色体对中的百万碱基大小的单链DNA片段随机分布到许多纳升的隔间中,在微流控装置中进行酶扩增,从而提高了测序的准确性。通过在反应中添加抗外切酶终止子,PTA技术产生更小的双链扩增产物,从而进行有限的后续扩增。这导致反应从指数过程转变为准线性过程,增加了来自主模板的扩增量,并增强了基因组扩增的覆盖率和均匀性。目前,基于MDA的产品已经成熟,如Qiagen公司的REPLI-g单细胞试剂盒。
MALBAC
Zong等人(2012)于2012年报道,MALBAC(多重退火和基于循环的放大周期)是一种scWGA方法,旨在减轻与非线性放大相关的偏差。MALBAC引物的5’端共有27个核苷酸序列,3’端有8个随机核苷酸序列。扩增过程始于这8个随机核苷酸与0°C下的基因组DNA模板杂交。然后,当温度升高到65°C时,我们使用具有链置换活性的DNA聚合酶来创建半扩增子不同的长度(0.5-1.5kb)。在94°C时,然后将半扩增子从模板中变性。半扩增子被进一步扩增,产生具有互补末端的完整扩增子,当温度降低(至58°C)时形成发夹。全扩增子形成环,可能会阻碍进一步的扩增和交叉杂交。经过5轮预扩增后,将含有常见的27个核苷酸序列的寡核苷酸作为PCR的引物,对整个扩增子进行指数扩增。这一过程产生了下一代测序所需的微克DNA。此外,基于MALBAC方法的商业试剂盒已经被创建,如MALBAC单细胞WGA测试(Qiagen)。
MALBAC不仅仅是DOP-PCR和MDA的组合;由于它的准线性放大,它具有根本的不同,它绕过了与指数放大相关的序列依赖性偏倚,从而增强了放大的均匀性。在MALBAC技术的初始阶段,采用多重置换反应,使扩增产物的全基因组覆盖率达到93%。MALBAC具有广泛的覆盖范围和均匀的扩增,使其适用于单个细胞中SNPs和CNVs的全基因组检测(Hou,2013;Zong,2012)。该技术的实施促进了临床辅助生殖技术的发展(Yao,2018)。此外,MALBAC在SNV检测中的假阴性率较低。然而,与MDA技术相比,MALBAC在SNV检测中确实表现出更高的假阳性率,因为目前使用的DNA聚合酶的假阳性率较低。
LIANTI
与以前的WGA方法,如指数PCR反应退化PCR((Telenius,1992),链取代DNA聚合酶驱动指数放大MDA单链DNA(Dean,2001),和准线性放大MALBAC(Zong,2012)通过循环扩增子保护和PCR都涉及非特异性引物和指数扩增导致偏差和错误,一种新的scWGA方法由Chen等的团队(Chen,2017),称为线性扩增通过转座子插入(LIANTI)。LIANTI利用Tn5转座酶技术在全基因组扩增过程中维持线性扩增。该方法将基因组DNA随机切片,然后用预先确定的序列填充切割位置通过利用Tn5转座酶的特性,LIANTI将T7启动子插入到基因组DNA中。然后,将标记有T7启动子的基因组DNA片段用T7 RNA聚合酶进行IVT,线性扩增成数千个RNA拷贝。接下来是RT和第二链合成,产生为DNA文库构建准备的双链LIANTI扩增子。与早期的技术(DOP-PCR、MDA和MALBAC)相比,LIANTI在识别CNVs和SNV方面分别表现出优越的灵敏度和准确性。此外,LIANTI优于其他WGA技术(DOP-PCR、MDA和MALBAC),其基因组覆盖率为97%,等位基因退出率为17%的WGA技术。
META-CS
真正的SNV必须位于两条DNA链上的相同位置,而聚合酶错误和DNA损伤通常随机发生在两条链中的一条上。因此,对双链DNA(dsDNA)的两个互补链进行测序,对于减少单链的假阳性和提高准确性至关重要。互补链的多重末端标记扩增(META-CS)是一种革命性的scWGA技术,由Xie及其同事于2021年提出(Xing,2021)。由于DNA的互补性,META-CS可以在单管反应中清晰地识别和扩增这两条DNA链,减少了几乎所有的假阳性。使用这种方法可以从单个细胞中可靠地识别从头SNV。
META-CS是建立在以前报道的多路复用的由本研究团队开发的末端标记放大方法。最初,将16个独特的转座子序列的组合与Tn5转座子酶以等摩尔比混合,形成转座子复合物。这些复合物从单个细胞中随机切割基因组DNA。随后,由两个随机转座子序列标记的基因组DNA片段通过加热进行变性,释放出两条单链。为了获得链特异性标记,这些链随后使用两个连续的聚合酶延伸过程进行预扩增。每次聚合酶延伸反应后,使用外切酶I来消除多余的引物。这些产物,从原始DNA的义链和反义链中分别扩增出来,可以通过将它们映射到参考基因组来区分。因此,SNV是由来自两条链的信息确定的,显著提高了准确性。
高通量scWGS方法
自从第一个用于分析人体组织拷贝数的scWGS方法的引入以来(Navin,2011),单细胞基因组学领域在过去的十年中取得了快速的进展。早期的技术仅限于一次分析少量细胞,并基于WGA化学物质(Chen,2017;Dean,2001;Telenius,1992)。然而,对肿瘤细胞基因组进化和重建的研究往往需要同时分析大量单细胞肿瘤基因组的突变特征。低通量的scWGS方法由于其劳动密集型、昂贵、耗时的性质和有限的效率而带来了挑战。组合索引、纳米管和微滴技术的最新发展极大地提高了细胞吞吐量并降低了成本(Laks,2019;Minussi,2021;Vitak,2017;Yin,2019)。在下面的章节中,我们将详细地讨论和比较基于不同策略的高通量scWGS方法。支持信息中的表S5总结了几个关键的高通量scWGS方法的特点。
基于微流控的高通量scWGS方法
(1) 直接库制备方法(DLP)
Zahn等人(2017)提出了一种直接DNA转位单细胞文库生产(DLP)方法。该方法直接从单个单元格中创建索引库。DLP方法利用一种特殊设计的微流控装置来捕获和溶解单个细胞。Tn5转座体复合物随机分割单个细胞的基因组DNA片段,在5‘端标记一个不同的适配器序列。然后,使用11个PCR周期,将索引条形码和测序适配器添加到标记的DNA插入物的两端。在索引之后,这些库被组合用于多路测序。
在建立文库之前,早期的单细胞技术使用WGA来捕获整个基因组(Gawad,2014;Navin,2011;Wang,2014)。然而,预扩增引入了扩增偏差,降低了覆盖一致性,阻碍了CNVs的检测(Macaulay和Voet,2014;Wang,2012;Zong,2012)。DLP是第一个直接生产而不使用标记进行预扩增的单细胞文库的一个例子。该方法产生高均匀覆盖的基因组,使许多细胞多路复用,使其适合高通量和价格合理的CNV检测。与DOP-PCR相比,DLP更具成本效益(每个细胞约0.50美元,而每个细胞15美元)和时间效率(2.5h vs 3d)。然而,微流控装置的使用限制了细胞的吞吐量,并需要一定大小的细胞,因为非常小的细胞可能会滑过陷阱,除非该设备是专门为这种类型的细胞制造的。大细胞也可能堵塞通道。因此,仍然需要额外的优化。
(2) 微流控液滴法
由于单细胞分配方法的局限性、从单细胞扩增基因组DNA的困难以及文库制备的酶学步骤的复杂性,提高细胞测序的吞吐量面临着挑战(Lan,2017;Vitak,2017)。为了应对这些挑战,Andor等人(2020年)提出了一种实现大规模scWGS的解决方案。他们的方法利用基于两阶段微流控液滴技术自动生成高细胞数的scWGS库。与之前的单细胞转录组研究类似,微流控液滴上装载了一个可以标记DNA的条形码水凝胶珠。为了制造细胞珠(CBs),单个细胞首先被包裹在水凝胶基质中。然后将用于细胞裂解和蛋白质消化的试剂添加到这些CBs中,以裂解和解开DNA。为了制造细胞珠-凝胶珠(CBGB)乳液,我们使用了第二个微流控芯片,其中凝胶珠(GB)被数百万个不同的液滴识别条形码副本功能化,并与水凝胶CB和酶反应混合物共封装。CBGB在封装后进行溶解,释放内容。用测序适配器和条形码序列标记的基因组DNA片段通过两步等温孵育过程获得。在破碎和纯化乳剂后,文库已经准备好进行Illumina测序。这种基于微流控液滴的细胞分离技术可以在一次实验中分离出数万个细胞,其吞吐量超过了传统的DLP技术。
基于纳米细胞的高通量scWGS方法
(1) 高通量直接换位scWGS方法(DLP+)
与基于预扩增的技术相比,早期使用微流控装置的DLP方法有效地减少了偏差(Zahn,2017)。尽管基于微流控的DLP分析性能良好,但定制的微流控设备的使用限制了细胞的大小,并对其普遍采用和可扩展性提出了障碍。此外,一些基于液滴的方法对细胞大小也面临着类似的限制(Andor,2020)。为了应对这些问题,Emma Laks(Laks,2019)开发了DLP+平台,这是一种高通量直接换位scWGS系统,建立在“现成的”皮升体积压电分配技术和商品高密度纳米管阵列上。在DLP+方法中,通过限制稀释分离单细胞,并分配到纳米细胞阵列中。为了达到几乎完美的单细胞分离率,所选择的细胞使用定位软件选择性地沉积到反应室中,该软件将它们定位在分配喷嘴内。一个10倍的倒置荧光显微镜扫描每个纳米细胞芯片,以验证单细胞的占用率,并收集有关细胞状态的数据。在发现文库制备试剂之前,要进行成像,使其能够在成像过程中排除双孔、空孔或受污染的细胞。添加试剂,旋转,密封,和加热芯片是程序参与从未扩增的单细胞中创建DLP+文库。使用标准的Illumina程序和HiSeq设备,得到的文库在恢复过程中被汇集,并在所需的覆盖深度进行测序。
通过其透明的分配喷嘴和内置相机,DLP+提供了一个独特的优势,使在分配前拍摄物体的高分辨率显微镜图像成为可能。通过积极选择单个细胞,这一功能有助于防止碎屑或双链细胞的测序。此外,通过有系统地修改一些变量,包括细胞裂解体积和缓冲类型、转座酶(Tn5)浓度、索引后PCR周期和细胞裂解/DNA溶解时间,DLP+改进了物理反应决定因素,以生产高质量的文库。DLP方法是这些优化的基础(Zahn,2017)。此外,DLP+显示出基于微流控与DLP方法相当的基因组覆盖一致性,但吞吐量明显更高,在一系列组织类型中,每次实验从数百到数万个细胞。
(2) 档案纳米细胞测序方法(Arc-well)
先前开发的高通量scWGS方法,包括DLP(Laks,2019)、SCI-seq(Vitak,2017)和ACT(Minussi,2021),有一个共同的限制——它们需要新鲜或快速冷冻的组织样本,使它们不适合分析档案福尔马林固定石蜡包埋(FFPE)组织样本。为了解决这一挑战,Wang等人(2023)引入了一种名为Arc-well(档案纳米细胞测序)的新方法。为了进行Arc-well,对FFPE块进行切片和脱亲和,产生单核悬液,随后用于FACS分类。将分类后的细胞核分布到5184孔的纳米孔芯片中,可以对纳米孔进行成像,以选择单细胞,防止双态、细胞核恶化和空孔。然后,使用ICELL8 cx系统(TaKaRa Bio)进行五步等量分配步骤,该系统用于将下游试剂分配到纳米细胞芯片中。首先,分配裂解试剂来裂解选定的细胞核并释放基因组DNA。接下来,分配用于标记反应(Tn5转座体)和Tn5失活。此外,通过沉积双指标(72个×72个组合)和扩增PCR结果,每个纳米孔都得到一个不同的条形码组合。然后将条形码库汇集在Illumina平台上进行排序。
声细胞标记(ACT)技术于2021年首次提出。该技术利用声学液体转移(ALT)技术、基因组DNA直接标记和单核流式细胞仪,可以实现单分子分辨率的高通量单细胞DNA测序(Minussi,2021)。将ACT方法与Arc-well进行比较,发现Arc-well具有更高的吞吐量(每次实验1900-2600个细胞),更低的试剂成本和更低的技术可变性。重要的是,Arc-well证明了扩增FFPE档案组织中常见的降解DNA片段的能力,使其与此类组织样本兼容。
基于组合索引的高吞吐量scWGS方法
(1) SCI-seq
FACS在SCI-seq方法中用于将单个细胞分类为96孔板(Vita,2017)。随后,来自单个细胞的基因组DNA被Tn5转座酶随机片段,每个产生的片段都用索引1和一个适配器标记。指数2的引入是通过PCR反应来实现的。最终,这些不同的文库被组合起来进行测序。核小体缺失被SCI-seq用于组合索引程序,这使得同时产生数千个单细胞基因组测序文库成为可能。这种方法除了具有高通量外,还具有不需要专门的微流体设备或液滴乳化程序的优点。然而,值得注意的是,SCI-seq技术在PCR扩增过程中引入了一定的偏倚。
(2) SCI-L3-WGS
为了解决放大偏差问题,Yin等人(2019)引入了sciL3,这是一种将组合索引与线性放大相结合的方法。在3级索引技术的帮助下,sciL3-WGS显著提高了LIANTI的吞吐量,允许它在每次实验中至少测序数千甚至数百万个细胞,同时最大限度地减少扩增偏差。sci-L3- WGS过程分为三个关键步骤:(i) Tn5转座酶从单个细胞中随机切割基因组DNA,并将条形码1连接到每个片段上。(ii)第二组条形码被连接到DNA片段的末端,以及一个位于两个条形码外的T7启动子。(iii)引入的T7启动子启动IVT,然后是RT和第二链合成。在第二链合成过程中引入了第三组条形码和UMIs。双链DNA分子可以按照传统的文库制备程序进行制备。每个分子包含三个识别细胞来源的条形码。与目前的方法和任何直接的SCI-seq(Vitak,2017)和LIANTI(Yin,2019)的组合相比,sci-L3策略有许多好处首先,使用IVT,它实现了与LIANTI相同的线性放大。其次,由于它使用了三轮条形码,其理论吞吐量超过100万个细胞,而文库制备成本低廉(Cao,2019)。第三,sci-L3是一种灵活的线性扩增结合高通量细胞索引的策略;除单测序外,还可用于其他单细胞测序分析,如单细胞RNA/ DNA共分析。
scWGS在生物医学中的应用
scWGS技术能够揭示单细胞基因组结构的差异,是一个强大的工具,被用于许多不同的领域,包括肿瘤生物学、体细胞突变和镶嵌现象、生物体发育、生殖细胞突变和发育、生育能力和微生物研究。它已成为生命科学的一个主要研究领域。该技术在生育和肿瘤生物学领域的应用将是接下来讨论的重点。
(1) 肿瘤生物学
肿瘤是一种多方面的和多样化的疾病,其特征是基因组不稳定和体细胞突变的积累,其瘤内异质性对个体化癌症医学提出了重大挑战。传统的大规模测序方法为癌症的基因组组成提供了有价值的见解;然而,他们往往忽视了肿瘤内部固有的异质性,导致了对疾病的不完整的描述。scWGS的出现已经被证明有助于克服这一限制。通过支持对个人的分析在单分子水平上,scWGS在癌症研究的各个方面都显示出了显著的潜力,如阐明肿瘤内的异质性、解释克隆过程的进化、理解侵袭和转移、研究循环肿瘤细胞(CTCs)和评估治疗结果。
一项针对乳腺恶性肿瘤的研究揭示了首次利用基于DOP-PCR的scWGS研究肿瘤内异质性。该研究利用拷贝数变化确定了乳腺肿瘤内的亚克隆谱系(Navin et al.,2011)。后续scWGS研究,利用多种癌症类型,如卵巢(McPherson,2016)、膀胱(Li,2012)、大脑(Francis,2014)、肾(Xu,2012)、结直肠(Leung,2017)、肝癌(Hou,2016)、肺(Ferronika,2017)和血液学(Gawad,2014;Hughes,2014)癌症扩大了我们对CNVs和SNV水平的瘤内异质性的理解。这些研究揭示了特定病例中肿瘤亚型和亚克隆多样性之间的相关性。例如,Baslan等人(2020年)使用基于DOP-PCR的测序方法,对16个乳腺癌样本中的2086个乳腺细胞基因组进行了全面分析。他们观察到,与雌激素受体阳性乳腺癌相比,雌激素受体阴性乳腺癌表现出更高的亚克隆多样性。
通过单细胞DNA测序(scDNA-seq)获得的基于肿瘤内异质性谱的系统发育分析,为识别在癌症发展和进展中发挥重要作用的驱动突变和基因改变提供了有价值的见解。通过分析单个癌细胞的基因组,研究人员能够确定驱动肿瘤生长的特定突变,为开发解决这些驱动突变的靶向治疗提供关键信息。在Wang等人(2014)的一项工作中,使用靶向双单分子测序和scWGS的组合对数百个乳腺细胞进行了分析。在两名乳腺癌患者中,研究人员研究了突变进化和克隆多样性。他们的研究表明,SNV逐渐进化,导致了高度的克隆多样性。另一方面,非整倍体重排发生在肿瘤发生的早期,并在克隆生长过程中保持非常稳定。该研究在雌激素受体阳性的浸润性导管癌样本中发现了许多与癌症相关的非同义突变,如PIK3CA、CASP3、FBN2和PPP2R5E。有趣的是,在腔内A乳腺肿瘤中最常见的驱动突变是PIK3CA(Ellis,2012;Network,2012)。
CTC起源于原发肿瘤并进入外周血,有可能促进转移。CTC的ScWGS为肿瘤的无创取样提供了一种很有前途的方法,为无创预后甚至诊断提供了见解。在Riebensahm等人(2019)的一项研究中,我们采用scWGS分析了乳腺癌脑转移患者CTC的基因突变特征。该研究确定了突变基因,如TP53、ARID1A、CDH1和TTN,与ARID1A一起参与染色质重构,被强调为一个潜在的药物靶点。Ni等人(2013年)采用了MALBAC方法,检测了从肺癌患者中获得的单个CTC的基因组。分析显示,在CTC外显子组中存在与癌症相关的插入/缺失(indels)和SNV。这一突变信息为个性化治疗提供了潜在的临床指导。此外,CTC已被用于治疗反应的无创监测(Dago,2014)。
总之,scWGS是癌症研究中的一项开创性技术,提供了对单个癌细胞基因组景观的全面理解。通过发现克隆进化、识别驱动基因突变、跟踪染色体异常、研究肿瘤微环境和检测微小残留疾病,scWGS为癌症诊断、预后和靶向治疗提供了有价值的见解。随着scWGS技术的不断发展,它为推进个性化癌症医学带来了巨大的前景。
(2) 生育能力
通过体外受精(IVF)产生的胚胎的植入前遗传学诊断(PGD)和植入前基因组筛选(PGS)是scWGS的两种临床用途。这有助于防止有害突变和染色体异常的遗传,使染色体的彻底研究。为此,使用了多种基因组分析系统,包括多重定量PCR、比较基因组杂交(CGH)阵列和SNP阵列(Rubio,2013;Tobler,2014;Treff,2012)。scWGS技术改进了分析胚胎活检的传统方法,能够同时识别整个基因组的非整倍体和突变(Kumar,2015;Treff,2013;Wells,2014)。高通量测序方法的快速发展进一步降低了费用,提高了PGD/PGS在染色体水平上的准确性和分辨率。这种方法有望提高在体外受精过程中选择健康胚胎的准确性,提高辅助生殖的成功率,并降低新生儿发生遗传疾病的风险。下面,作者将描述PGD/PGS中scWGS的几个应用程序示例。
在PGS和PGD中scWGS的应用已经在许多研究中得到证实:Wells等人(2002)利用基于pcr的WGA在第一极体上进行scWGA,使用CGH技术成功检测胚胎的染色体异常。Daina等人(2013)对一个Lynch综合征家族的14个胚胎进行了单基因分析,采用MDA方法成功实现了双因素PGD,并导致了2名健康儿童的出生。Hou等人(2013)采用基于MALBAC的测序技术,分析了来自8个健康供体的单个人类卵母细胞的基因组。他们展示了如何在体外受精过程中通过MALBAC PGS选择正常受精卵进行胚胎移植。Huang等人(2014)收集了3名孕妇供体的23个冷冻切割胚胎,并进行了单细胞CGH、SNP和MALBAC测序,并进行了24条染色体的非整倍体分析。MALBAC测序结果与CGH和SNP的一致性较高,表明其在PGD/PGS中具有应用价值。Shang等人(2018)将MALBAC-scWGS扩展到应用于线粒体疾病的PGD/PGS检测,证明了该技术在解决各种遗传条件方面的多功能性。
ScWGS通过分析胚胎内的单个细胞,使PGD/PGS检测发生了革命性的变化。这种强大的技术提供了关于染色体异常、结构变异和突变景观的详细信息。检测胚胎内单个细胞的基因组含量的能力提高了遗传分析的精度,为选择具有最高水平的胚胎提供了有价值的见解在体外受精期间成功的可能性。因此,scWGS有助于提高体外受精程序的成功率。
总结
单细胞基因组学技术的进展并没有跟上转录组学的步伐,这主要是由于在DNA捕获过程中实现甚至是基因组覆盖的挑战。尽管如此,单细胞基因组测序已经为各种以前无法触及的生物学问题带来了重要的见解。该技术在不同的研究领域都有应用,包括体细胞突变、了解基因组功能、研究生物体发育和探索微生物学。单细胞基因组测序在临床和翻译研究和实际应用方面显示出巨大的潜力,特别是在肿瘤学和辅助生殖领域。
参考文献:
Sun F, Li H, Sun D, et al. Single-cell omics: experimental workflow, data analyses and applications. Sci China Life Sci. 2025;68(1):5-102. doi:10.1007/s11427-023-2561-0